This is the most common life-limiting inherited disease in Caucasians.
It is caused by a genetic mutation that alters mucous production, resulting in a more viscous solution.
It is autosomal recessive.
Life expectancy is improving. Newborns with CF have a life-expectancy of about 40 years.
The condition is inherited in an autosomal recessive pattern.
Epidemiology
- Most common in Caucasians, rare in other races.
- In Caucasians, 1 in 25 people are carriers, and 1 in 2500 births have CF.
o In Asians the risk is only 1/10 of this: 1 in 25 000.
Pathology and Genetics
- CF is the result of mutations on the long arm of chromosome 7, in the material that codes for the Cystic fibrosis transmembrane regulator (CFTR) protein. This is essentially a chloride channel. There are many identified mutations, but the most common is known as ΔF508, and accounts for 80% of cases.
o Parents with different mutations can still have a child with CF. Having a mutation basically means that 50% of the CFTR’s you produce will be defective (CF only becomes symptomatic at 3% of normal function). So, if you have a child with another carrier, the actual mutation is insignificant – you will have a ¼ chance of having a child with two defective genes for CFTR.
- On mucosal surfaces, this channel normally allows chloride ions out of the cell in the presence of cAMP, and into the lumen (e.g. of the airway or pancreas).
o Cystic fibrosis will result when CFTR activity is less than 3% of normal. A less severe form of the condition, sometimes call pancreatic sufficient CF will occur when CFTR activity is 3-8% of normal.
- The result of this reduced chloride level is an increased re-absorption of sodium from the fluid in the lumen. This, in turn, causes a reduced excretion of water.
- The viscous mucous plugs the exocrine ducts of the pancreas
o Note that in sweat glands, the CFTR plays a different role in ion regulation – allowing the reabsorption of chloride ions from the sweat – hence the sweat test (see below) for CF.
Clinical features
- CLASSICAL PRESENTATION – a child age 0-2:
o Recurrent Infections
o Large offensive stools
o Failure to thrive
Breakdown of symptoms:
- Newborn
o Malabsorption and failure to thrive from birth
o Recurrent / persistent chest infections, which often produced purulent sputum.
§ The nature of the mucous (thick, not easily passed up the mucociliary escalator, thus there is stasis of mucus) predisposes to chronic infection with organisms such as S Auerus and H influenzae, and later in life Psuedomonas Aeruginosa
§ These chronic infections combined with the chronic inflammatory state, causes bronchiectasis and abscess formation.
o Meconium Ileus – 10-20% of cases - meconium is the substance that is present in the intestine at birth. It is dark, sticky and odorless, and has an appearance like tar. It is made of bile, intestinal cells, water, amniotic fluid and has a high concentration of mucous. Normally, after birth, pancreatic enzymes act on the meconium, and it is passed in the stools. In meconium ileus the meconium is not broken down by the pancreatic enzymes due to pancreatic insufficiency. As a result, it may cause obstruction. This can be treated with Gastrografin enema, or oral N-acetylcysteine,but in many cases will require surgery.
o In the UK – screening of newborns is performed
- Age 0-2
o All of the above, plus:
§ Extended period of neonatal jaundice
§ Steatorrhoea
- Age 2-8
o All of the above, plus:
§ Bronchiectasis
§ Rectal prolapse
§ Nasal Polyps
§ Sinusitis
- Age 8+
o All of the above, plus:
o Cor pulmonale
o Diabetes Mellitus (often NIDDM) – ultimately occurs in 25% of patients. Tends to develop in adolescence or later, and is a result of declining pancreatic function.
o Cirrhosis and portal hypertension. May also be hepatomegaly.
§ Liver transplant may be considered in some cases, and is generally very effective.
o Distal intestinal obstruction (similar to meconium ileus)
o Pneumothorax
o Haemoptysis
o Infertility – in males – due to lack of vas deferens. Fertility in females normal. Men can still father children via intracytoplasmic sperm injection.
o Psychological disorders
o Cyanosis
o Clubbing
o Osteoporosis – thought to be related to nutrition
- Other Features
o 90% of CF patients have pancreatic insufficiencies. This leads to maldigestion and absorption, which is the cause of the failure to thrive in many infants. When pancreatic insufficiency is present there will usually be large, pale, smelly stools. These stool also predispose to rectal prolapse.
§ In CF pancreatic secretions are rich in mucous and protein. This means they are particularly viscous, and can form plugs in the pancreatic ductules. This then allows the enzymes to act on the pancreatic acinar cells, eventually destroying them.
- Examination
o Chest hyperinflation – resulting from air trapping
o Inspiratory crepitations
o Expiratory wheeze
§ Ask the patient to wheeze/cough if they are able
o Hepatomegaly
o Clubbing (after several years)
o Check for nasal polyps
o Cyanosis
o Signs of resp infection
Investigations
Diagnostic:
- Sweat test – this uses electrodes to provoke sweating. Two electrodes are placed on the skin, and a small current passed through them. Filter paper is used to collect a sample of the sweat. Then the concentration of both sodium and chloride ions is measured:
o Child - >60 mmol/L – is abnormal
o Adult - >90mmol/L is abnormal
§ Chloride usually > sodium
§ The test can be inaccurate if not enough sweat is collected, and is highly operator dependent.
- Elastase in faeces – low levels of elastase in the faeces is diagnostic for pancreatic insufficiency.
- Screening in newborns – has been shown to improve lung function in the child. Tests include:
o Guthrie test – Immunoreactive Trypsin (IRT) is raised in CF babies. If this test is positive, the babies are screened for common CF mutations, and have a sweat test to confirm the diagnosis.
- Other tests – probably not performed for diagnosis, but abnormal results may be spotted on routine investigations:
o GTT – raised
o Vitamins A, D, E, K – low
o CXR – hyperinflation, bronchiectasis
o Abdominal ultrasound – fatty liver, cirrhosis, chronic pancreatitis
o Spirometry – obstructive pattern
Management
As always, is best done with an MDT. Very important to involve the parents as part of the team as well. Patients should be followed-up in outpatients at least once a year.
There is no cure for CF. The aims of treatment are to minimize the progression of the disease, and maintain adequate nutrition and development.
- Nutrition – there is a very strong link between good nutrition and prognosis for CF.
o Oral pancreatic enzymes are taken by most CF patients. They are used not only to allow digestion and absorption of food, but also to control steatorrhoea and stool frequency.
o PPI’s – e.g. omeprazole are also often given to provide the optimal duodenal pH for the oral enzymes to function
o CF patients also require a high energy diet, to compensate for the reduced digestion. It is recommended that patients have 120-150% the normal recommended intakes. Fat intake should not be restricted - steatorrhoea can be controlled by the levels of oral pancreatic enzymes. It is also important to take supplemental fat-soluble vitamins (A, D, E, K).
o NG or gastronomy tube feeding may be used in some patients to supplement normal intake methods to ensure high enough calorie intake.
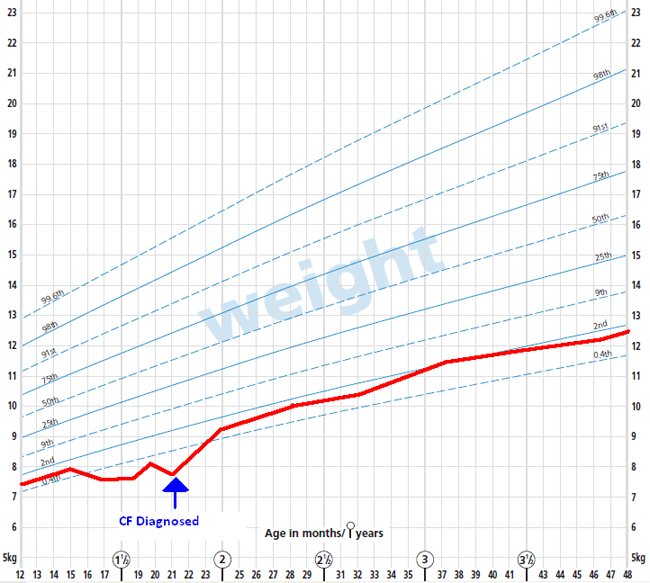
o Growth – you should aim for normal growth. Often the child may fall behind the percentiles before the diagnosis, but then recover, and follow normal percentiles after diagnosis and management has been instigated. E.g.:
- Respiratory complications – the treatment for the respiratory problems associated with CF is very similar to that of chronic bronchiectasis.
o Antibiotic therapy
§ Many CF patients use oral antibiotics; often a prophylactic agent (e.g. flucloxacillin) is used, and another agent added in acute exacerbations. As infection become more chronic and established, they may resist therapy.
· Some experts believe that prophylactic antibiotics become far less effective after just 3-4 months of use. Current recommendations do not require cycling of antibiotics (Which has been proven to reduce resistance), but it is likely that in the future the use of prophylaxis will be more selective, and will involve regular cycling (e.g. every 6 weeks) of antibiotic agents to reduce resistance.
§ Nebulised agents may be given between exacerbations to try and suppress long standing infections, particularly pseudomonas. IV agents can also be used, again typically against pseudomonas, and many CF patients will be trained to self-administer this at home. Ultimately it is likely that patients will be permanently colonized with highly resistant strains.
· Pseudomonas infection is associated with worse prognosis and is especially difficult to treat. By adulthood, most patients have a chronic pseudomonas infection, with a unique strain of pseudomonas. These can be especially virulent, and CF patients are advised not to mix with oneanother – to avoid the risk of contracting another’s strain.
· Asperigillus and other unusual mycobacteria are also often colonizers of CF patients, although there clinical role is thought to be minimal.
§ Mucolyitcs – e.g. DNases – these are products that break down DNA found in the sputum as a result of severe breakup of inflammatory cells. These drugs can help to make the mucus less viscous. This can both improve pulmonary function, and reduce the number of exacerbations.
§ Azithromycin is another antibiotics that is often used. It has been proven to reduce the frequency of exacerbations, however, this is probably the result of immunomodulatory actions, and not of a direct antibiotic effect.
o Nebulisers – may be useful in some patients, especially those with symptoms consistent with asthma, although a diagnosis of asthma is often difficult to confirm.
§ Nebulised Saline has also been shown to reduce the number of infective exacerbations.
o Home oxygenation and CPAP – non-invase oxygen therapies may be used in the later stages of the disease
o Lung transplant – can dramatically improve function, and may be considered later in life. It is very rare in children, and for adults there are not enough donors to supply demand.
- Postural drainage
o Patient positioned so the trachea is inclined downwards, allowing gravity to help the flow of mucous out of the lungs. With the aid of physiotherapy and ‘chest percussion’ (clapping on the patients back with a cupped hand), this can help to dislodge mucous. A typical physiotherapy regimen may be 3x/day for 1-20 minutes.
§ This can help completely drain the chest of mucus, and many children can lead symptom free lives with the right treatment.
- Liver complications
o Ursodeoxycholic acid (UDCA)– is given for the prevention of liver impairment.
§ Up 20% of adolescents with CF will develop liver complications. Ursodeoxycholic acid is sometimes used in primary biliary sclerosis and gallstones as it alters the metabolism of cholesterol and micelles, and makes them break down more readily. The evidence for its use in CF is patchy, but it is thought to reduce cholestasis and aid bile acid reabsoprtion, which help increase liver function in individuals with CF.
o Liver transplant – may be suitable for some patients with severe cirrhosis.
Prognosis
- 95% of patients will die from respiratory complications.
- Respiratory infections are very common during childhood.
- Disease progression can be measured with spirometry – using FEV1 numbers measured against predicted levels for height and weight. A relative low FEV1 gets worse at the disease progresses.
- Respiratory failure is the main cause of death – usually during an acute exacerbation of resistant infection.
Future
Future treatments could involve gene transfer therapy, whereby the affected CTFR gene could be replaced by a fully functioning version through adenovirus therapy. CF is a particularly good candidate for such therapy, as the bronchial mucosa is easier accessible via inhalation.
Notes by Tom Leach
EmoticonEmoticon